
Apert Syndrome – Acrocephalosyndactyly
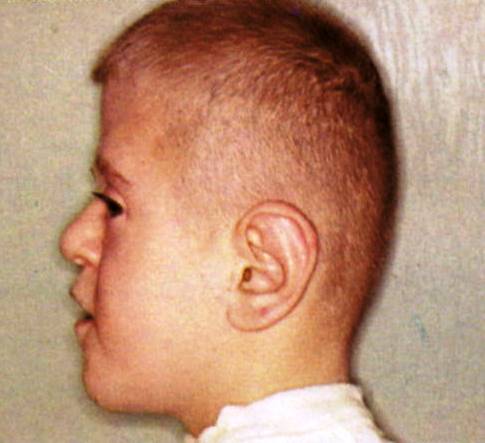
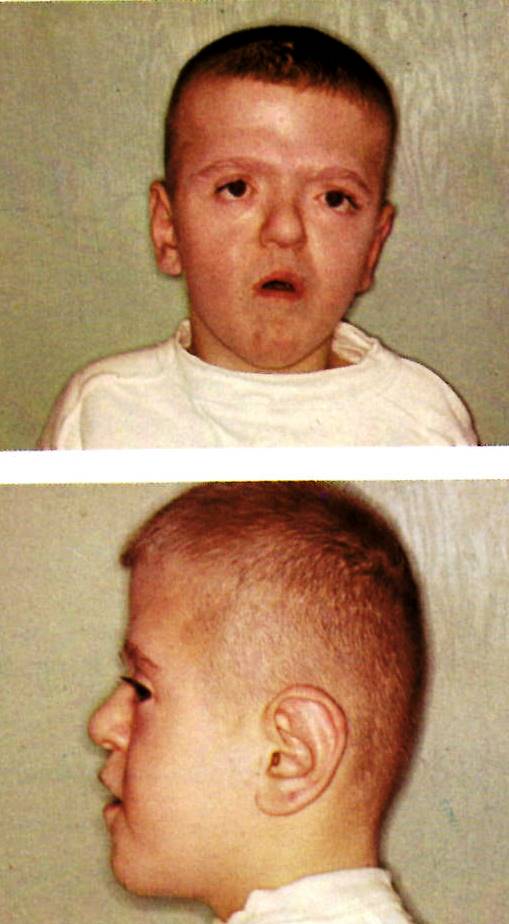
Illustrated is a young male with a combination of ACRO-CEPHALO-SYNDACTYLY (ACS). Originally, attention was called to this concurrence of connective tissue anomalies in 1906 by Eugene Apert. Since then it is known that several gene mutations can manifest as ACS and distinguished by other concurrent anomalies.
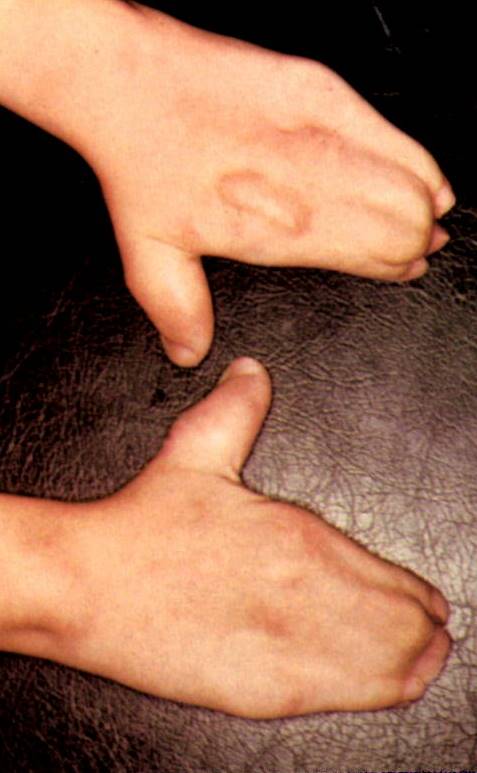
Turricephaly, the most stigmatic sign in combination with syndactyly is strongly diagnostic. The turricephaly is usually associated with coronal suture fusions and therefore in a lengthening of the calvarium vertically.Facial synostoses are also present and the most obvious impacts are on the maxillae and in severe instances manifested as bulging eyes (exophthalmos which requires attention) associated with a downward or antimongoloid slant of the palpebral fissures. The ears and nose tend to be small. The oral cavity is reduced in size resulting in crowded teeth, nasal obstruction and cleft palate. Regarding the limbs, as mentioned in the comments about the images of the hand, the syndactyly and the presence or not of polydactyly are critical signs to identify subtypes of ACS. In these syndromes synostosis and ankylosis of other bones and impacts on articulations (joints) are common.
Many instances are associated with intellectual deficits.
ACS type 1 or Apert syndrome is due to an autosomal gene mutation, mostly sporadic but if not, resulting in dominant phenotypes (in hetero zygotes, presumably, homozygosity is lethal). Reproduction by two affected individuals in unlikely.
This observation is an example of convergence of multiple etiologies upon connective tissue development and modifying impacts by the genome on the general clinical phenotype (polymorphism). Clinically, one primary classification is to distinguish if there is presence of polydactyly – in this case, there is not. This patient may represent one of four ACS categories (gene mutations): Apert (mostly, FGFR2 mutation); Crouzon (mostly, also FGFR2 mutation); Saethre-Chotzen (a variety of mutations); and Pfeiffer (also FGFR2 mutation).
My personal impression is that this patient represents ACS type 1 or Apert syndrome.
CREDIT: The images and clinical profile are mostly adapted from “Atlas of Mental Retardation Syndromes” by U.S. Department of Health, Education, and Welfare. 1968 (0-310-072 U.S. Government Printing Office), edited by S. S. Gellis, M.D. and M. Finegold, M.D.
201708161747